Fenilcetonuria: signos clásicos de transmisión hereditaria y terapia dietética
Contenido
- 1¿Cómo se manifiesta la fenilcetonuria?
- 2Mecanismo de desarrollo de la enfermedad
- 3Fenilcetonuria en niños
- 4Síntomas de la enfermedad
- 5Causas y desencadenantes
- 6Diagnóstico
- 7Tratamiento de la fenilcetonuria clásica
- 8Características de la nutrición de los recién nacidos ydietoterapia
- 9Dieta para niños en edad preescolar y escolares
- 10Grupos de productos con PKU
- 11Cómo controlar el nivel de fenilalanina en la sangre
- )12Video
Las enfermedades cuya aparición está relacionada con defectos en el aparato celular genético - fenilcetonuria - se incluyen en una pequeña lista de enfermedades hereditarias que son tratables.El pionero de esta dolencia fue un médico noruego IA Felling, más tarde se descubrió que un solo gen llamado gen de fenilalanina hidroxilasa (el brazo largo del duodécimo cromosoma que contiene hasta 4.5% de todo el material de ADN celular) fue responsable del desarrollo y el curso de la enfermedad.El defecto hereditario conduce a la desactivación parcial o completa de la enzima hepática fenilalanina-4-hidroxilasa.
Cómo se detecta la enfermedad de fenilcetonuria
La enfermedad de fenilcetonuria hereditaria (PKU) conduce a una intoxicación crónica del cuerpo con sustancias tóxicas formadas como resultado del metabolismo y el proceso de aminoácidos deterioradoshidroxilación de fenilalanina.La intoxicación permanente causa daño al sistema nervioso central (SNC), cuya manifestación es una disminución progresiva de la inteligencia (oligofrenia fenilpirúvica).
La enfermedad de Felling se manifiesta en la acumulación excesiva de fenilalanina en el cuerpo y en los productos de su metabolismo incorrecto.Otros factores en el desarrollo de fenilcetonuria incluyen transporte de aminoácidos deteriorado a través de la barrera hematoencefálica, recuentos bajos de neurotransmisores (serotonina, histamina, dopamina).En ausencia de un tratamiento oportuno de la enfermedad, se produce un retraso mental y puede causar la muerte del niño.

El mecanismo del desarrollo de la enfermedad
La causa de los trastornos genéticos es un bloqueo metabólico que impide la formación de fenilalanina-4-hidroxilasa (una enzima,que es responsable de la conversión del aminoácido fenilalanina en tirosina).El aminoácido proteinógeno tirosina es una parte integral de las proteínas y el pigmento de melanina, por lo que es un elemento necesario para el funcionamiento de todos los sistemas del cuerpo, y su falta conduce a la fermentopatía.
La inhibición de la formación de metabolitos causada por la inactivación mutacional de la enzima es la activación de las vías metabólicas auxiliares de la fenilalanina.El alfa-aminoácido aromático, como resultado de procesos metabólicos defectuosos, se divide en derivados tóxicos, que en condiciones normales no forman:
- ácido fenilpirúvico (fenilpiruvato) - un ácido alfa-ceto graso-aromático, su formaciónconduce a la mielinización de procesos neuronales y demencia;
- el ácido fenil láctico es un producto formado durante la recuperación del ácido fenilpirúvico;
- feniletilamina: el compuesto inicial para transmisores biológicamente activos de impulsos electroquímicos, aumenta la concentración de dopamina, adrenalina y noradrenalina;
- El acetato de ortofenilo es una sustancia tóxica que causa trastornos metabólicos en el cerebro.
Las estadísticas médicas indican que un gen alterado patológicamente está presente en el 2% de la población, pero no se manifiesta de ninguna manera.El defecto genético se transmite al niño de los padres solo en presencia de la enfermedad en ambos cónyuges, y el bebé en el 50% de los casos se convierte en el portador del gen mutado, permaneciendo sano.La probabilidad de que la fenilcetonuria en los recién nacidos provoque la enfermedad es del 25%.
Por qué tipo se hereda
La enfermedad de Felling es una anomalía genética heredada de un tipo autosómico recesivo.Este tipo de herencia significa que el desarrollo de signos de una enfermedad congénita solo ocurrirá cuando una copia genética defectuosa de ambos padres que son portadores heterocigotos del gen alterado se herede del niño.
El desarrollo de una enfermedad congénita en el 99% de los casos es causado por una mutación del gen responsable de codificar la enzima que proporciona la síntesis de fenilalanina-4-hidroxilasa (fenilcetonuria clásica).Hasta el 1% de las enfermedades genéticas están relacionadas con cambios mutacionales que ocurren en otros genes que causaninsuficiencia de dihidropteridina reductasa (PKU tipo II) o tetrahidrobiopterina (PKU tipo III).
Fenilcetonuria en niños
La forma clásica de enfermedad genética en niños en la mayoría de los casos se manifiesta externamente, comenzando a los 3-9 meses de edad.Los recién nacidos con un gen defectuoso, se ven saludables, una característica distintiva es el hábito específico (apariencia) del bebé.La sintomatología expresada aparece en 6-12 meses después del nacimiento.
La PKU tipo II se caracteriza por el hecho de que los primeros síntomas clínicos aparecen 1,5 años después del nacimiento.Los signos de la enfermedad no desaparecen después del diagnóstico de anomalías genéticas y el comienzo de la terapia dietética.Este tipo de enfermedad congénita a menudo conduce a un desenlace fatal durante 2-3 años de la vida de un niño.Los síntomas más comunes de la PKU tipo II son:
- trastornos mentales graves;
- hiperreflexia;
- alteración de la función motora de todas las extremidades;
- síndrome de contracciones musculares no controladas.
Los signos clínicos de cambios mutacionales en los genes tipo III son similares a los de la enfermedad tipo II.La deficiencia de tetrahidrobiopterina se caracteriza por una tríada de síntomas específicos:
- un alto grado de retraso mental;
- el tamaño del cráneo se reduce claramente en relación con otras partes del cuerpo;
- espasticidad muscular (es posible la pérdida completa de la movilidad de las extremidades).

Manifestaciones de la enfermedad de Felling
En ensayos clínicos y observaciones, se ha sugerido queLos derivados tóxicos del metabolismo de la fenilalanina causan una disminución de la capacidad intelectual, que es de naturaleza progresiva y puede conducir a la demencia (oligofrenia, idiotez).Entre las causas probables de trastornos irreversibles de la actividad cerebral de los más razonables se considera que es causada por una disminución en el nivel de tirosina falta de neurotransmisores que transmiten impulsos entre las neuronas.
La relación exacta de causa y efecto entre la enfermedad hereditaria y los trastornos cerebrales aún no se ha identificado, al igual que el mecanismo de desarrollo debido a la fenilcetonuria de estados mentales como la equopraxia, la ecolalia, los ataques feroces y la irritabilidad.Los resultados del análisis indican que la fenilalanina tiene un efecto tóxico directo en el cerebro, que también puede causar una disminución en la inteligencia.
Estructura y características fenotípicas
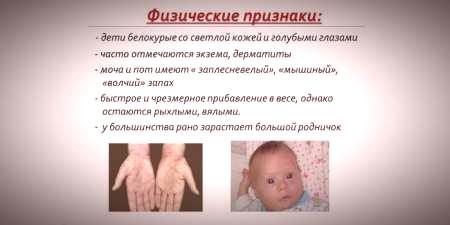
Dado que la saturación de pigmento de la piel y el cabello depende del nivel de tirosina en las mitocondrias de hepatocitos, y la fenilcetonuria conduce a la interrupción de la conversión de fenilalanina, los pacientes con esta enfermedad tienen peculiaridadessignos recesivos).El aumento del tono muscular provoca la aparición de desviaciones en la estructura del cuerpo: se vuelve displásico.Las características externas distintivas de la fenilcetonuria incluyen:
- hipopigmentación: piel clara, ojos azul pálido, cabello descolorido;
- cianosis de extremidades;
- tamaño de cabeza reducido;
- posición específica del cuerpo: cuando trata de pararse o sentarse, el niño adopta una postura "a medida" (brazos y piernas doblados en las articulaciones).
Síntomasenfermedad
Con la detección oportuna, la enfermedad de Felling se trata con éxito ajustando la nutrición y el desarrollo del niño ocurre de acuerdo con su grupo de edad.La dificultad de detectar una mutación genética es que los signos tempranos son difíciles de detectar incluso por un pediatra experimentado.La gravedad de los síntomas de la enfermedad congénita aumenta a medida que el niño crece, porque el consumo de alimentos proteicos contribuye al desarrollo de trastornos del SNC.
Signos en los recién nacidos
Durante los primeros días de vida de un niño, los signos de anormalidades anormales son difíciles de detectar: el bebé se comporta naturalmente, no hay retrasos en el desarrollo.Los síntomas de la enfermedad comienzan a aparecer por primera vez 2-6 meses después del nacimiento.Los padres deben estar atentos al comportamiento del bebé, que se caracteriza por una baja actividad, letargo o, por el contrario, ansiedad, hiper excitabilidad.
Con el inicio de la lactancia materna, los recién nacidos con leche comienzan a ingresar al cuerpo del recién nacido con leche, que es un catalizador para la aparición de los primeros signos, que indican claramente que la enfermedad ha comenzado a progresar.Las manifestaciones clínicas específicas de la enfermedad incluyen:
- vómitos constantes (a menudo considerado estrechamiento congénito del portero);
- vómitos frecuentes;
- ninguna respuesta a estímulos externos;
- distonía muscular (tensión muscular reducida);
- síndrome convulsivo (convulsiones de naturaleza epiléptica o no epiléptica).
Síntomas en niños después de 6 meses
Si la manifestación de enfermedad genética no esocurrió (o no se observó) durante los primeros 6 meses después del nacimiento del niño, luego de este período ya es posible determinar con precisión el retraso en el desarrollo psicomotor.Los síntomas de trastornos genéticos causados por deficiencia enzimática en niños mayores de seis meses son:
- disminución de la actividad (hasta la indiferencia completa);
- falta de autocontrol, asientos;
- un olor peculiar de la piel a "ratón" (el olor a moho resulta de la excreción de derivados de fenilalanina tóxicos a través de las glándulas sudoríparas y la orina);
- pérdida de la capacidad de reconocer visualmente los rostros de los padres;
- descamación de la piel;
- dermatitis, eccema, esclerodermia.
Progresión de la enfermedad en ausencia de tratamiento en la infancia
Si no se detectaron anomalías del desarrollo en la infancia y no se llevó a cabo el tratamiento adecuado, entoncesla enfermedad comienza a progresar activamente y a menudo conduce a discapacidad.La falta de terapia en una etapa temprana de la enfermedad hace que se desarrollen los siguientes síntomas a la edad de 1,5 años:
- microcefalia (disminución del tamaño del cerebro);
- prognatia (desplazamiento de la dentición superior hacia adelante);
- dentición tardía;
- hipoplasia del esmalte (adelgazamiento o ausencia total de esmalte dental);
- la demora en el desarrollo del lenguaje hasta la ausencia total del habla;
- 3, 4 grados de oligofrenia (retraso mental, retraso mental);
- defectos cardíacos congénitos (defectos en la estructura del músculo cardíaco, partes del corazón,vasos grandes);
- trastornos del sistema autónomo (acrocianosis, sudoración excesiva, hipotensión arterial);
- estreñimiento.
Causas y factores provocadores
Para la manifestación de una mutación autosómica recesiva, se debe heredar un gen defectuoso de ambos padres.Las enfermedades genéticas de este tipo ocurren con la misma frecuencia en niños y niñas recién nacidos.La patogénesis de la PKU es causada por un metabolismo alterado de la fenilalanina, que puede presentarse en 3 formas.La terapia dietética se somete solo a fenilcetonuria clásica tipo I.
Las formas atípicas de la enfermedad pueden curarse ajustando la nutrición.Estas desviaciones son causadas por una deficiencia de tetrahidropterina, deshidropterina reductasa (raramente - piruviltetrahidropterina sintasa, guanosina-5-trifosfato y otros).La mayoría de los casos de casos fatales se registran entre pacientes con variaciones raras de PKU, y las manifestaciones clínicas de todas las formas de la enfermedad son similares.El riesgo de tener un bebé con un gen de fenilalanina hidroxilasa mutada aumenta si los padres son parientes cercanos (en matrimonios estrechamente relacionados).
Diagnóstico
En caso de sospecha de trastornos genéticos, el diagnóstico se realiza sobre la base de un conjunto de datos obtenidos del estudio del historial médico: datos genealógicos, los resultados de estudios genéticos clínicos y médicos.Para la detección oportuna de enfermedades congénitas (PKU, fibrosis quística, galactosemia, etc.) se desarrolló un programa obligatorio de masasExamen de laboratorio de todos los recién nacidos (cribado neonatal).
Si los futuros padres conocen el portador del gen mutado, la medicina moderna ofrece formas de detectar un defecto en la etapa del embarazo (diagnóstico prenatal del feto por método invasivo).Para la división de fenilcetonuria en tipos por gravedad, se utiliza una clasificación condicional basada en el nivel de fenilalanina en un fluido de plasma sanguíneo libre de fibrinógeno:
- Fenilcetonuria pesada - 1200 μmol /l.
- Promedio - 60-1200 µmol /l.
- Luz (no requiere tratamiento) - 480 μmol /L.
Prueba de detección
La detección de anormalidades genéticas ocurre en varias etapas.En la primera etapa en el hospital de maternidad, se toman muestras de sangre periférica (de cinco) a todos los bebés de 3-5 días de vida para la investigación.El material se aplica en papel y se envía al laboratorio bioquímico donde se realiza el análisis bioquímico.En la segunda etapa de la prueba de detección, se determina la concentración del valor normal de fenilalanina.

Si no se detectan cambios patológicos, se completa el diagnóstico y se registra la tarjeta del niño.En caso de desviaciones de la norma, los resultados del diagnóstico se envían al pediatra para garantizar un examen más preciso de la muestra de sangre del recién nacido.La salud del niño depende de la implementación oportuna y precisa de todas las medidas para detectar desviaciones.Si el diagnóstico se confirma después de la detección repetida, los padres del niñoenviado a una clínica de genética pediátrica para recibir tratamiento.
Análisis y estudios para confirmar el diagnóstico
El nuevo diagnóstico tras la detección durante la prueba de detección primaria de anomalías se realiza al volver a enviar las pruebas.Además de determinar el contenido de fenilalanina en la sangre a los métodos de diagnóstico de PKU en niños y adultos, se incluyen:
- Prueba de tala: determinación del ácido fenilpirúvico en la orina mediante la adición de cloruro de hierro al biomaterial (tinción en color azul-verde);
- Prueba de Guthrie: evaluación del grado de respuesta de los microorganismos al metabolismo o enzimas contenidos en la sangre del paciente;
- cromatografía: el estudio de las propiedades químicas de las sustancias distribuidas entre dos fases;
- fluorimetría: irradiación de biomaterial por radiación monocromática para determinar la concentración de sustancias contenidas en el mismo;
- electroencefalografía: diagnóstico de actividad eléctrica del cerebro;
- La resonancia magnética es la excitación de los núcleos atómicos de las células por ondas electromagnéticas y la medición de su respuesta.
Tratamiento de la fenilcetonuria clásica
En el corazón de la terapia con fenilcetonuria hay una restricción en el consumo de productos que son la fuente de proteínas animales y vegetales.El único método de tratamiento exitoso es la terapia dietética, cuya idoneidad se evalúa por el contenido de fenilalanina en el suero.Nivel máximo de aminoácidos en pacientes de diferentes grupos de edad.es:
- en bebés y niños de hasta 3 años, hasta 242 µmol /l;
- en preescolares de hasta 360 µmol /l;
- en pacientes de 7 a 14 años: hasta 480 μmol /l;
- en adolescentes: hasta 600 μmol /l.
La efectividad de la dieta depende de la etapa de la enfermedad para la que se corrige la dieta.En el diagnóstico temprano de patología congénita, la terapia dietética se prescribe a partir de la octava semana de vida (después de este período, ya comienzan los cambios irreversibles).La ausencia de medidas oportunas conduce a complicaciones y una disminución del nivel de inteligencia en 4 puntos durante 1 mes desde el nacimiento hasta el comienzo del tratamiento.

Teniendo en cuenta que la dieta terapéutica para la fenilcetonuria proporciona la exclusión completa de la dieta de la proteína animal, es necesario utilizar otras fuentes de aminoácidos esenciales, así como vitaminas del grupo B, calcio.y compuestos minerales que contienen fósforo.Los productos que se utilizarán como suplementos sin proteínas incluyen:
- hidrolizados de proteínas (Amigen, Aminazol, Fibrinosol);
- no contienen mezclas de fenilalanina saturadas con aminoácidos esenciales - Tetrafen, libre de fenilo.
Además de las medidas curativas para eliminar la causa del deterioro del funcionamiento del cuerpo, se debe llevar a cabo un tratamiento sintomático destinado a eliminar los defectos del habla y normalizar la coordinación de los movimientos.La terapia compleja incluye procedimientos de fisioterapia, masajes, la ayuda de un terapeuta del habla, un psicólogo, la realización de ejercicios de gimnasia.En algunos casos, junto con la terapia dietética.muestra el uso de anticonvulsivos, fármacos nootrópicos y vasculares.
Peculiaridades del tratamiento de formas atípicas
La fenilcetonuria tipo II y III no se puede tratar con una dieta baja en proteínas: el nivel de fenilalanina en la sangre permanece sin cambios mientras se limita el flujo de proteínas al cuerpo o los síntomas clínicos progresan incluso con la disminución en el nivel de aminoácidos.La terapia efectiva de estas formas de la enfermedad se lleva a cabo con el uso de:
- tetrahidrobiopterina - factor de la enzima afectada;
- análogos sintéticos de tetrahidrobiopterina: estas sustancias penetran mejor en la barrera hematoencefálica;
- fármacos de terapia de sustitución: no eliminan la causa de la fenilcetonuria, pero apoyan el funcionamiento normal del cuerpo (Levodopa junto con Carbidofa, 5-oxitriptófano, 5-formiltetrahidrofolato);
- hepatoprotectores - apoyan la función hepática;
- anticonvulsivos;
- la introducción del gen de fenilalanina hidroxilasa en el hígado es un método experimental.
Nutrición del recién nacido y terapia dietética
La lactancia materna está permitida en el primer año de vida de un bebé con PKU, pero debe restringirse.Hasta 6 meses, la ingesta aceptable de fenilalanina es de 60-90 mg por 1 kg de peso para bebés (100 g de leche contienen 5,6 mg de fenilalanina).A partir de los 3 meses, la dieta del bebé debe ampliarse gradualmente, introduciendo jugos y purés de frutas.
Los niños a partir de los 6 meses de edad pueden ingresar a la dieta purés de verduras, gachas (sagú), sin proteínasácidoDespués de 7 meses, es posible darle al bebé pasta baja en proteínas, a partir de los 8 meses, pan, que no contiene proteínas.No se ha establecido la edad a la que se debe restringir la proteína en el cuerpo del niño enfermo.Los médicos aún discuten la viabilidad de la terapia dietética de por vida, pero están de acuerdo en que se debe seguir una dieta de al menos 18.
La fenilcetonuria diagnosticada en una mujer no es una razón para rechazar el nacimiento de un niño.Las futuras madres con PKU para prevenir lesiones fetales durante el embarazo y para evitar posibles complicaciones, es necesario observar una dieta con una dieta restringida en fenilalanina (en el momento del parto) (hasta 242 μmol /l).
Mezclas sin lactosa para bebés
La dieta para la fenilcetonuria se basa en una reducción significativa en la cantidad de proteína natural en la dieta diaria, pero el cuerpo del bebé no puede desarrollarse normalmente en ausencia de los oligoelementos necesarios.Para satisfacer las necesidades de proteínas del bebé, se utilizan mezclas de aminoácidos sin lactosa que, según la ley rusa, deben proporcionarse de forma gratuita.
La tolerancia del bebé a la fenilalanina cambia rápidamente durante el primer año de vida, por lo que debe controlarse su concentración en la sangre del bebé y ajustarse la dieta.Las mezclas están diseñadas para ciertos grupos de edad:
- bebés menores de un año se prescriben Afenilak 15, Analog-SP, PKU-1, PKU-mix, PKU Anamix;
- Niños mayores de 1años, nombrar mezclas enriquecidas con vitaminas y minerales con alto contenido de proteínas: PKU Prima, P-AM Universal, PKU-1, PKU-2, Maximeid XP, Maximum XP.

Alimentos con suplementos proteicos
Uno de los componentes principales de una dieta para la fenilcetonuria son los productos a base de almidón bajos en proteínas.Estos suplementos contienen hidrolizado de caseína, triptófano, tirosina, metionina, nitrógeno y proporcionan el requerimiento diario del bebé de la proteína necesaria para el desarrollo y el crecimiento normales.Los alimentos especializados que llenan la falta de minerales y aminoácidos esenciales cuando faltan en la dieta son:
- Berlofeno;
- Zimorgan;
- Minafen;
- Aponti.
Dieta para preescolares y escolares
A medida que el cuerpo se adapta a la fenilalanina, los niños a partir de los 5 años pueden reducir gradualmente sus restricciones dietéticas.La expansión de una dieta ocurre por la introducción de cereales, productos lácteos, productos cárnicos.Los estudiantes mayores ya tienen una alta tolerancia a la fenilalanina, por lo que a esta edad es posible continuar la expansión de la dieta, con la necesidad de controlar la respuesta a cualquier cambio en la nutrición.Los siguientes métodos se utilizan para controlar la condición del niño:
- evaluación de parámetros neurológicos, estado psicológico;
- control del rendimiento del electroencefalograma;
- determinación del nivel de fenilalanina.
Grupos de alimentos con PKU
La dieta de los pacientes con PKU, junto con baja en proteínasLos productos con almidón y las mezclas medicinales también incluyen productos de origen natural.Al elaborar el menú, la cantidad de proteína consumida debe calcularse claramente y la dosis recomendada por el médico no debe exceder.Para eliminar los efectos tóxicos en el cuerpo, se han desarrollado 3 listas de productos que contienen artículos prohibidos (rojo), no recomendados (naranja) y permitidos (verde).
Lista roja
La fenilcetonuria se desarrolla en el contexto de la ausencia de una enzima que se convierte en tirosina fenilalanina, por lo que un alto contenido de proteínas es la base para incluir los productos en la lista (roja) prohibida.Los elementos de esta lista deben excluir por completo la dieta de un paciente con PKU:
- carne;
- órganos internos de animales, subproductos;
- salchichas, salchichas;
- mariscos (incluido el pescado);
- huevos de todas las aves;
- productos lácteos;
- nueces;
- legumbres y cereales;
- productos de soja;
- platos que contienen gelatina;
- productos de confitería;
- aspartamo.
Lista naranja
Los productos que se dosificarán a un niño diagnosticado con PKU se incluyen en la lista naranja.La inclusión en la dieta de los elementos de esta lista es aceptable, pero en un número estrictamente limitado.Aunque estos productos no contienen mucha proteína, también pueden aumentar el nivel de fenilalanina, por lo que no se recomienda su uso:
- vegetales enlatados;
- platos de patata y arroz;
- repollo;
- leche;
- sorbete.
Lista Verde
Los productos libres de proteínas pueden usarse en pacientes con diagnóstico de fenilcetonuria sin restricción.Antes de comprar los artículos de la lista verde, es necesario examinar la composición indicada en el paquete y asegurarse de que no contenga un tinte para el aspartamo que contenga fenilalanina:
- frutas;
- hortalizas (excepto papas y coles);
- bayas;
- verduras;
- cereales con almidón (sagú);
- miel, azúcar, mermelada;
- productos a base de harina de maíz o harina de arroz;
- aceites, grasas (mantequilla, girasol, oliva).
Cómo controlar el nivel de fenilalanina en la sangre
La fenilcetonuria es una enfermedad incurable que puede transferirse a la fase de estancamiento mediante el uso de una dieta y medidas terapéuticas.Con el cambio de las condiciones de vida, la alteración de la dieta, la enfermedad puede agravarse nuevamente, por lo tanto, los pacientes necesitan observación de por vida.El proceso de control consiste en determinar periódicamente el nivel de fenilalanina en la sangre.La frecuencia del análisis depende de la edad del paciente:
- hasta 3 meses: se debe realizar un análisis de sangre semanalmente para obtener resultados consistentes;
- de 3 meses a 1 año - 1-2 veces al mes;
- 1 a 3 años, una vez cada 2 meses;
- mayores de 3 años - trimestralmente.

La sangre para análisis se administra 3-4 horas después de la ingestión.Además de la detección, el desarrollo de la PKU se controla determinando el estado nutricional, el desarrollo físico y emocional del paciente, el nivel de habilidades intelectuales.y desarrollo del lenguaje.Las observaciones pueden requerir diagnósticos adicionales con la participación de especialistas apropiados.
Video
La información presentada en este artículo es solo orientativa.El artículo no exige autotratamiento.Solo un médico calificado puede diagnosticar y recomendar un tratamiento basado en las características individuales del paciente en particular.